The APyT spectrum fitting command line script
The fitting of mass spectra obtained with the apyt_spectrum_align script is
based on an analytic model describing the generic peak shape. All peaks in the
spectrum are represented using a shared set of peak shape parameters, with only
a single intensity parameter required per isotopic peak group (see also the
spectrum fitting module). This approach makes
extensive use of the Python periodic table module.
Note
A major advantage of this fitting approach is that it supports not only individual atoms but also molecules. Provided the mass spectrum is properly aligned and all species with their respective charge states are correctly specified, spectra with more than 100 peaks can typically be fitted reliably.
Invoking the command line script
The script apyt_spectrum_fit is installed automatically when following the
installation instructions. It requires two positional
arguments:
Measurement ID — the record ID in the database.
Species dictionary — a Python dictionary specifying the chemical species and charge states present in the spectrum.
To see all available options, run:
apyt_spectrum_fit --help
Commonly used options include:
--no-sql— Skip connecting to an SQL database. Instead, load measurement data and metadata from a local database. This is the typical mode for local testing or standalone workflows.--cache— Load measurement data from a binary NumPy.npyfile in the working directory. This avoids repeated file parsing or database queries, significantly speeding up subsequent runs.Note
The cache file is created automatically on the first run and reused for subsequent runs on the same measurement ID.
Note
This option only takes effect when the measurement data is retrieved from the SQL database.
The species dictionary must be given as a Python-style dictionary string. Each entry has the following format:
'<symbol>': ((<charge_states>), <atomic_volume_nm3>)
<symbol>: chemical symbol (used as key). This can be either an element or a molecule.<charge_states>: tuple of charge states to fit.<atomic_volume_nm3>: atomic volume in nm³ (used later in reconstruction).
For example, a tungsten spectrum (only triply charged, volume 0.0158 nm³) can be provided with:
apyt_spectrum_fit --no-sql 1 "{'W': ((3,), 0.0158)}"
Tip
For complex spectra, specifying the properties dictionary directly on the
command line can be cumbersome. Instead, you can store the dictionary in a
custom file, e.g. my_species.ppy:
{
'W': ((3,), 0.0158)
}
and pass it to the script using:
apyt_spectrum_fit --no-sql 1 "<(cat my_species.ppy)"
This makes the invocation clearer and easier to maintain.
Graphical user interface
Running apyt_spectrum_fit opens an interactive graphical interface for
spectrum fitting. The example below shows a tungsten reference measurement:
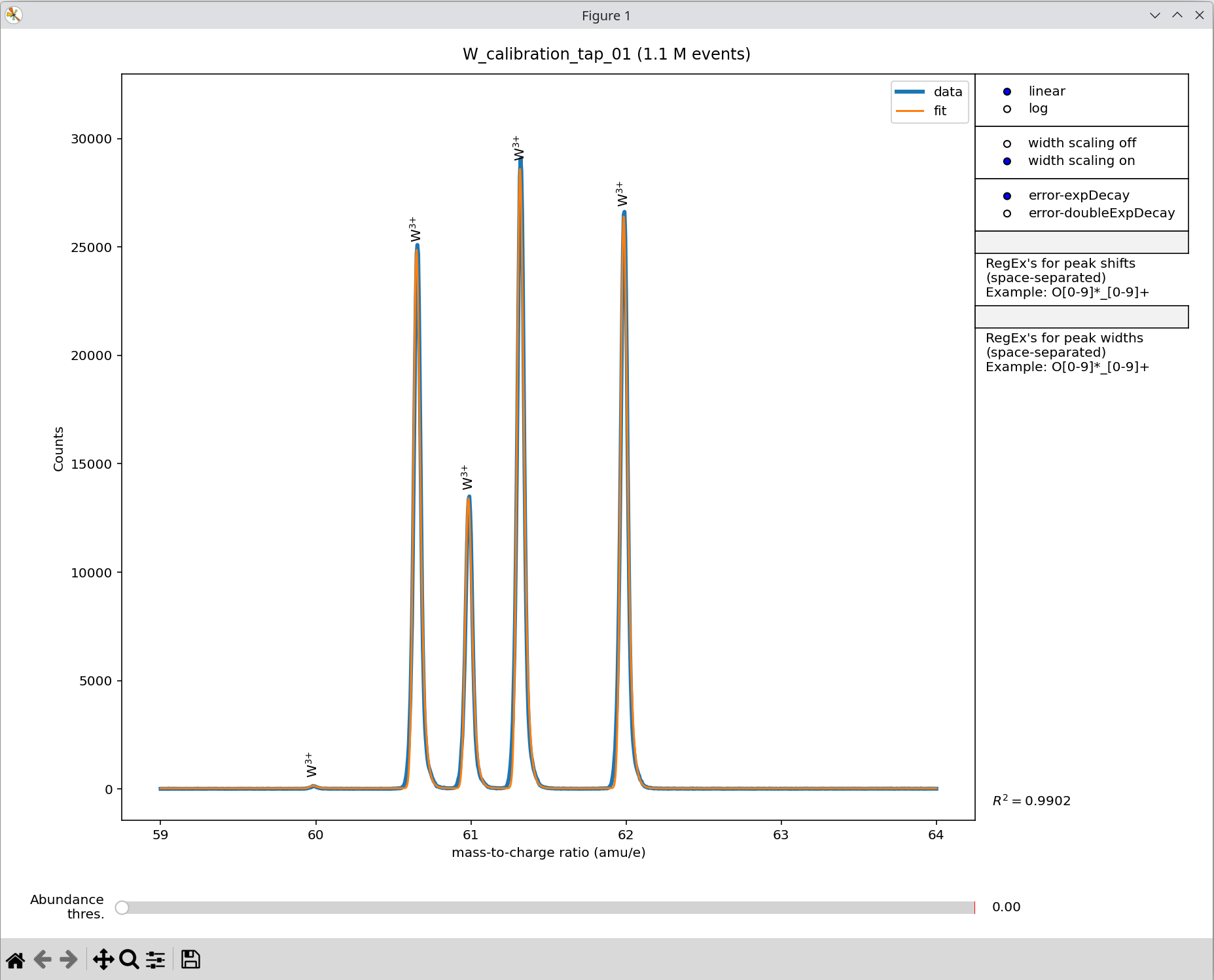
Exemplary mass spectrum fitting for a tungsten measurement using the
apyt_spectrum_fit command line script.
The interface consists of:
A main panel showing the experimental mass spectrum (blue) and fitted spectrum (orange).
A slider (Abundance thres.) at the bottom for controlling automatic peak labeling.
Toggles and input fields on the right-hand side.
The right-hand controls include:
linear/log — Toggle between linear and logarithmic scaling of the spectrum.
width scaling off/on — Enable an additional free fitting parameter that scales peak width with mass-to-charge ratio. By default, a square-root scaling (exponent = 0.5, as expected physically) is used, but experimental data often shows broader peaks.
error-expDecay / error-doubleExpDecay — Choose the analytic peak-shape function. Both are error-function onsets with one or two exponential decays describing peak tailing, respectively.
RegEx’s for peak shifts — In rare cases, certain species appear systematically shifted in the spectrum (e.g., oxygen in oxides). Additional shift parameters can be applied to peaks that match a regular expression.
Note
In the description shown, all pure oxygen peaks (single O atoms or On molecules with arbitrary charge state) are shifted together using the regular expression
O[0-9]*_[0-9]+. The latter part of this expression specifies the charge states.Multiple regular expressions can be provided, separated by spaces.
Attention
Use these regular expressions only if it is confirmed that certain peaks are genuinely shifted in the spectrum. This option should not be used to correct or “heal” a misaligned mass spectrum.
R² value (bottom right) — Coefficient of determination, a measure of the fit quality.
During fitting, additional details on species intensities, abundances, and composition are reported in the console.
See also
For technical details and implementation notes, see the spectrum fitting module.